D. Michalski,1 M. Heindl,1,2 J. Kacza,3 F. Laignel,1,2 L. Küppers-Tiedt,1 D. Schneider,1 J. Grosche,2 J. Boltze,4,5 M. Löhr,6 C. Hobohm,1 W. Härtig2
1Department of Neurology, University of Leipzig; 2Paul Flechsig Institute for Brain Research, University of Leipzig; 3Department of Anatomy, Histology and Embryology, Faculty of Veterinary Medicine, University of Leipzig; 4Fraunhofer Institute for Cell Therapy and Immunology, Leipzig; 5Translational Centre for Regenerative Medicine, University of Leipzig; 6Department of Neurosurgery, University of Würzburg, Germany
Correspondence: Dr. Dominik Michalski, Department of Neurology, University of Leipzig, Liebigstr. 20, 04103 Leipzig, Germany.
Tel. +49.341.9724256 - Fax: +49.341.9724199.
E-mail: dominik.michalski@medizin.uni-leipzig.de
Key words: macrophages, microglia, astroglia, experimental stroke, tPA, HBO.
Acknowledgements: the authors would thank Dr. Petra Madaj-Sterba and Ms. Sigrid Weisheit (Medizinisch-Experimentelles Zentrum, University of Leipzig, Germany) for animal care and breeding. Ms. Ute Bauer (Paul Flechsig Institute for Brain Research, University of Leipzig, Germany) is gratefully acknowledged for excellent technical assistance.
Contributions: LKT, DM, CH, JB, underlying study design; DS, funding facilitation; LKT, DM, animal experiments conduction; MH, FL, WH, tissue preparation and serial staining performing; JK, semiquantitative analyses; JG, laser scanning microscopy; DM, data analysis and manuscript writing; WH, JB, MH, ML, manuscript critical revision.
Conflict of interest: the authors declare no conflict of interest.
Received for publication: 11 December 2011.
Accepted for publication: 3 February 2012.
©Copyright D. Michalski et al., 2012
Licensee PAGEPress, Italy
European Journal of Histochemistry 2012; 56:e14
doi:10.4081/ejh.2012.e14
AbstractInflammation following ischaemic stroke attracts high priority in current research, particularly using human-like models and long-term observation periods considering translational aspects. The present study aimed on the spatio-temporal course of macrophage-like cell accumulation after experimental thromboembolic stroke and addressed microglial and astroglial reactions in the ischaemic border zone. Further, effects of tissue plasminogen activator (tPA) as currently best treatment for stroke and the potentially neuroprotective co-administration of hyperbaric oxygen (HBO) were investigated. Rats underwent middle cerebral artery occlusion and were assigned to control, tPA or tPA+HBO. Twenty-four hours, 7, 14 and 28 days were determined as observation time points. The accumulation of macrophage-like cells was semiquantitatively assessed by CD68 staining in the ischaemic area and ischaemic border zone, and linked to the clinical course. CD11b, ionized calcium binding adaptor molecule 1 (Iba), glial fibrillary acidic protein (GFAP) and Neuronal Nuclei (NeuN) were applied to reveal delayed glial and neuronal alterations. In all groups, the accumulation of macrophage-like cells increased distinctly from 24 hours to 7 days post ischaemia. tPA+HBO tended to decrease macrophage-like cell accumulation at day 14 and 28. Overall, a trend towards an association of increased accumulation and pronounced reduction of the neurological deficit was found. Concerning delayed inflammatory reactions, an activation of microglia and astrocytes with co-occurring neuronal loss was observed on day 28. Thereby, astrogliosis was found circularly in contrast to microglial activation directly in the ischaemic area. This study supports previous data on long-lasting inflammatory processes following experimental stroke, and additionally provides region-specific details on glial reactions. The tendency towards a decreasing macrophage-like cell accumulation after tPA+HBO needs to be discussed critically since neuroprotective properties were recently ascribed to long-term inflammatory processes. |
Ischaemic stroke is associated with an enormous socio-economic burden, but unfortunately only restricted treatment strategies are currently available.1 In recent years, the translation of preclinical successful attempts into the clinical setting has commonly failed, exemplified by the promising drug NXY-059.2 As one of the main consequences, an extended perspective of tissue salvaging was developed, which involves explicitly more than only neurons.1,3,4 The neurovascular unit represents a frequently used construct in this regard describing the interactions between neurons, vessels, astrocytes and associated microglia.5,6 Damaging events after ischaemic stroke caused by reduced cerebral blood flow due to an artery occlusion include mechanisms of hypoxia-related acute excitotoxicity, which overlap with long-lasting inflammatory processes, peaking in delayed stroke phases.7,8 Thereby, investigating the role of recruited immunoactive cells in stroke-affected areas has become a major interest in stroke research.3 Focussing on this topic, several studies have identified invading leukocytes and activated microglial cells as contributors with predominantly deleterious roles in acute stages after cerebral ischaemia.8,9,10,11,12,13,14,15 Further characterization of these cells also based on immunolabelling with antibodies directed against CD68, CD11b and the ionized calcium binding adaptor molecule 1 (Iba), but the identification of specific subpopulations remains a diagnostic challenge.16,17,18 Additionally, the spatio-temporal profile of macrophage infiltration in stroke-related brain tissue has been investigated in vivo by magnetic resonance imaging in rats,19 mice and humans.20,21 Taking these time-dependent changes into account, it becomes clear that experimental studies in the field of stroke should rely on long-term observation periods and should be based on rodent stroke models with pathophysiology comparable to the human condition, such as thromboembolic stroke.4,22 Unfortunately, these issues have often been neglected by previous studies.
Despite extensive research in the field of acute ischaemic stroke, tissue plasminogen activator (tPA) is currently the only drug-related treatment that has shown conclusive beneficial effects in the clinical setting.1,4,23 Nevertheless, also toxic effects of tPA have been reported, contributing to vascular disruption with potential cerebral haemorrhage caused by a tPA-mediated activation of matrix metalloproteinases (MMPs).24,25,26,27 In general, MMPs are known for their deleterious effects in acute stroke phases, but also beneficial properties in later phases have been discussed recently.24,28,29 In this context, invading leukocytes and local microglial cells were found to produce pro-inflammatory mediators,30,31,32,33,34 and act as primary source for MMP-9.29,35,36,37,38 Considering the deleterious effects of tPA influencing the immune-related response via MMPs, the idea of combined treatment strategies arose to attenuate harmful effects selectively.1,24
Hyperbaric oxygenation (HBO) represents the application of 100% oxygen at an elevated ambient pressure, which increases the rate of physically dissolved oxygen in the blood; hereby, the arterial oxygen pressure enhances from about 90 mmHg at room air to approximately 2000 mmHg while breathing 100% oxygen at 2.5 atmosphere absolute (ata).39,40 HBO has attracted attention in stroke therapy since preclinical studies have shown beneficial effects, e.g., concerning neurobehavioural testing and infarct size,41,42,43 as well as the frequency of secondary haemorrhages due to tPA.44 With regard to inflammatory processes, HBO was found to decrease the infiltration of neutrophils when administered prior to or immediately after permanent experimental cerebral ischaemia,45 and prior to reversible focal ischaemia.46 However, also a contrary report exists, which found no effect of HBO on neutrophil accumulation when administered after permanent experimental stroke.47 In recent years, HBO was discussed as treatment strategy in addition to tPA.48,49 In a first approach with simultaneous application of tPA and HBO, our group has demonstrated that combined treatment results in early clinical improvement, but did not observe consistent long-term effects following experimental stroke.50
The present study aimed on the long-term course of inflammatory processes after focal cerebral ischaemia in rats and considered tPA as widely used treatment as well as simultaneous HBO as a potential neuroprotective strategy in a translational relevant setting. For this purpose, we first addressed different regions of ischaemia with special emphasis on the spatio-temporal accumulation of macrophage-like cells and explored the link to the respective clinical course. Second, delayed cellular reactions, particularly micro- and astrogliosis in ischaemic border regions, were investigated considering the neurovascular unit concept to obtain further insights in long-term post-stroke processes. The emerging results might be useful in adapting future treatment strategies in stroke.
The study protocol involving animals has been proven by local authorities (Regierungspräsidium Leipzig, Germany) and was conducted according to the European Communities Council Directive (86/609/EEC). The brain tissue for the present histology-based investigation originated almost exclusively from our previous study.50 Herein, male Wistar rats – provided by the Medizinisch-Experimentelles Zentrum of the University of Leipzig (Germany) or Charles River (Sulzfeld, Germany) – underwent focal cerebral ischaemia by thromboembolic right-sided middle cerebral artery occlusion (MCAO). The animals were allocated to the following treatment groups: Control, tPA and tPA+HBO. Survival times of the animals after stroke induction were 24 h, 7, 14 and 28 days with subsequent sacrificing in deep narcosis. After perfusion with saline and 4% phosphate-buffered paraformaldehyde, brains were immediately removed from the skull and post-fixed with the same fixative for 24 h. Afterwards, the brains were equilibrated with 30% buffered sucrose and 30 µm-thick serial coronal forebrain sections were cut using a frozen microtome. All sections were collected in 0.1 M Tris-buffered saline, pH 7.4 (TBS) containing sodium azide. To allow semiquantitative analysis in the present study, tissue was selected according to the best-intact structures in combination with clearest infarct presentation, which resulted in a sample size of n = 3 for each treatment group at 24 h, 7, 14 and 28 days.
MCAO was induced according to a thromboembolic model, originally described by Zhang et al.51 with some modifications as described previously.50 Briefly, after cervical preparation of the right-sided arterial vessels, a polyethylene (PE) tubing with tapered end was inserted into the external carotid artery and moved forward into the internal carotid artery up to the origin of the middle cerebral artery. Here, a blood clot was injected followed by catheter removal and ligation of the external carotid artery stump. A further PE tubing that was inserted into the femoral vein served as secure access path for the future administration of tPA. During the surgical procedure, the animals were anesthetized with 2-2.5% isoflurane in a mixture of 70% N2O and 30% O2, and the body temperature was adjusted to 37.0°C by using a thermostatically controlled heating pad with a rectal probe (Fine Science Tools, Heidelberg, Germany).
In the control and tPA group, animals breathed room air at normal ambient pressure. Animals received tPA (Actilyse, Boehringer Ingelheim, Ingelheim, Germany) intravenously in a dose of 9 mg per kg body weight over 30 min with a total volume of 1 mL - starting 2 h after initiation of MCAO. Combined treatment with tPA and HBO was performed in a special chamber (Sayers/Hebold, Cuxhaven, Germany) with 100% O2 at 2.4 ata for a period of 1 h (excluding compression and decompression time); thereby, the administration of tPA was started simultaneously to HBO 2 h after initiation of MCAO.
Overall, CD68 was used for the identification of macrophage-like cells.16,18 Thereby, 8 sections per animal (containing the infarct region) were rinsed with TBS and endogenous peroxidase activity of tissues was abolished by treatment with 0.6% H2O2 for 30 min. Following several rinses with TBS, sections were blocked with 5% normal goat serum in TBS (+0.3% Triton X-100) for 1 hour. Subsequently, the tissue was incubated with mouse-anti-CD68 (AbD Serotec, Oxford, UK; 1:400 in the blocking solution) overnight. Next, the sections were extensively rinsed with TBS and processed with highly purified biotinylated goat-anti-mouse IgG (Dianova, Hamburg, Germany; 1:500 in TBS containing 2% bovine serum albumin = TBS-BSA) for one h. Following several rinses with TBS, the sections were applied to preformed streptavidin/ biotinyl-peroxidase complexes (12.5+5 µg/mL TBS-BSA) and then stained with diaminobenzidine/ammonium nickel sulphate as chromogen according to Härtig et al.52 Following all histochemical procedures, sections were briefly washed with distilled water, mounted on fluorescence-free glass slides, air-dried and coverslipped with Entellan in toluene (Merck, Darmstadt, Germany).
Considering that microglial and astrocytic activation are closely connected with inflammatory processes like neutrophil infiltration,10,13,32,53,54,55,56 multiple fluorescence labelling was performed for further characterization (see also Härtig et al.57). Generally, CD68 and CD11b were used for the identification of macrophage-like cells, whereas CD11b accents more granulocytes and neutrophils but also microglia; Iba predominantly labels microglia, while glial fibrillary acidic protein (GFAP) stains a large subset of astrocytes.16,18,32,58,59,60 According to the neurovascular unit concept, which describes amongst others the interaction of astrocytes and associated microglia with neurons,6,10 antibodies directed against neuronal nuclei (NeuN) were additionally used as neuronal marker.61 Table 1 provides an overview of the applied antibodies and triple immunofluorescence staining methods.
The omission of primary antibodies in control experiments led to the expected absence of any cellular staining. Switching the fluorophores related to the relevant markers resulted in unaltered staining patterns of CD68, CD11b, Iba, GFAP and NeuN.
An Axioplan microscope (Zeiss, Jena, Germany) was utilised for initial light and fluorescence microscopic analyses. Fluorescently labelled sections were also analysed with a confocal laser-scanning microscope (LSM) 510 Meta (Zeiss), equipped with an argon laser (488 nm) for the excitation of FITC and Cy2, and two helium-neon lasers for the excitation of Cy3 (543 nm), as well as Cy5 (633 nm). The following band-pass (BP) filters were used: BP 500-530 nm (Cy2, FITC, green), BP 565-615 nm (Cy3, red) and BP 654-718 nm (Cy5, infrared, colour-coded in blue). Original images were processed with Adobe Photoshop 7.0 (Adobe Systems Inc., Mountain View, CA, USA). Thereby, brightness, contrast and sharpness of some final pictures were slightly enhanced.
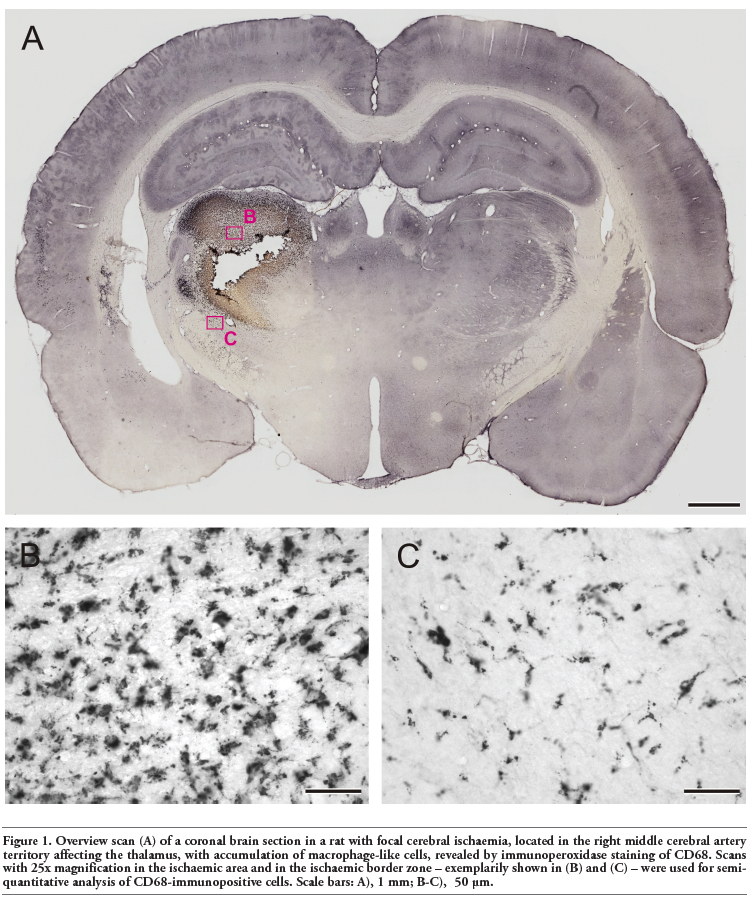
Sections of CD68-immunoperoxidase staining were used for semiquantitative analysis. Thereby, the section presenting the largest ischaemic infarct as well as the neighbouring rostral and frontal section (3 in total) were processed by using an Axioplan 2 imaging microscope (Zeiss) with digital camera (ORCA-ER, Hamamatsu, Hamamatsu City, Japan) and evaluation software (Volocity 4.3.2, PerkinElmer, Waltham, MA, USA). After orientation on an overview scan, one region of interest (ROI; dimensions 327.4x249.4 µm) was inserted into the ischaemic area, and a further ROI was placed in the ischaemic border zone. Using a 25x objective (numerical aperture 0.8), the CD68-immunoreactivity was analysed by intensity measurements of the whole area in each ROI, resulting in an amount of CD68-positive pixels per optic field as surrogate for the accumulation of macrophage-like cells. Thereby, the following settings were applied: find objects by standard deviation intensity (lower limit -100, upper limit -0.5) and exclude objects by size (<5 µm). Figure 1 shows a representative overview scan with exemplarily inserted ROIs. This overview was created by automatic scanning single images (Openlab 5.5.0, PerkinElmer) with a 10x objective and subsequent image stitching (Autopano Giga 2.0, Kano, France). Measurements were performed for each of both ROIs per section resulting in 6 values in total. Finally, means were calculated for both ROIs, which led to 2 values per study subject.
To explore the interrelation between the accumulation of macrophage-like cells and the clinical course, a linear regression model was calculated by using IBM SPSS Statistics (Vers. 20; IBM Corp., New York, NY, USA). Thereby, the ratio of CD68-positive pixel of the ischaemic area vs ischaemic border zone - representing the amount of cell accumulation - served as independent factor. In parallel, the change of the neurological deficit during the overall observation period served as dependent factor. The respective data on neurological impairment were extracted from our previous work,50 restricted to the subjects used in the present study.
The used embolic stroke model expectedly resulted in ischaemic infarctions with different morphologies primarily located in the middle cerebral artery territory involving the striatum, and partially affected the thalamus and hypothalamus. In general, macrophage-like cells were easily identified in ischaemic areas based on sufficient CD68-immunoperoxidase staining, which allowed a semiquantitative evaluation. Based on the observed accumulation of macrophage-like cells, an ischaemic core with maximal tissue damage and in later phases nearly complete loss of cellular staining could be distinguished from an ischaemia-related area with significant macrophage-like accumulation and from a surrounding ischaemic border zone with less macrophage-like cell accumulation (Figure 1).
|
Figure 1. Overview scan (A) of a coronal brain section in a rat with focal cerebral ischaemia, located in the right middle cerebral artery territory affecting the thalamus, with accumulation of macrophage-like cells, revealed by immunoperoxidase staining of CD68. Scans with 25x magnification in the ischaemic area and in the ischaemic border zone – exemplarily shown in (B) and (C) – were used for semiquantitative analysis of CD68-immunopositive cells. Scale bars: A), 1 mm; B-C), 50 μm. |
Figure 2 shows the time course of CD68-immunoreactivity in relation to the respective treatment and region of stroke-affected tissue. In the ischaemic area (Figure 2A), only a few macrophage-like cells were seen in all 3 groups at 24 hours after MCAO, followed by a drastic accumulation up to 7 days. At this time point, subjects of the control and tPA+HBO group displayed a tendency to a slightly higher macrophage-like cell accumulation when compared with tPA. Interestingly, the tPA+HBO group tended to less CD68-immunoreactivity in the ischaemic area during the following 3 weeks, whereas subjects of the control and tPA group provided similar courses. Markedly different findings were obtained in the ischaemic border zone (Figure 2B). During the first 14 days after MCAO, the accumulation of macrophage-like cells was found here at an almost constant low level. At 28 days, only a slight increase of CD68-immunoreactivity was observed in tPA-treated animals. In contrast, subjects of the tPA+HBO group tended to the lowest rate of macrophage-like cells in the ischaemic border zone.
Clinical relevance of macrophage-like cell accumulation
Based on the fact that CD68-immunoreactivity was markedly enhanced in the ischaemic area compared with its border zone (Figure 2), a pixel ratio between both areas was calculated as a surrogate for the accumulation of macrophage-like cells, and its association to the overall clinical course was further investigated. Figure 3 displays the relationship between both variables in terms of scatter plots addressing the overall sample (Figure 3A) and treatment-related populations, respectively (Figure 3B-D). Although statistical significance is generally lacking, we found a trend towards an association of accumulating macrophage-like cells and pronounced reduction of the neurological deficit during the observation period, indicated by negative Beta coefficients. This effect was most evident in the control group (Figure 3B), barely missing statistical significance.
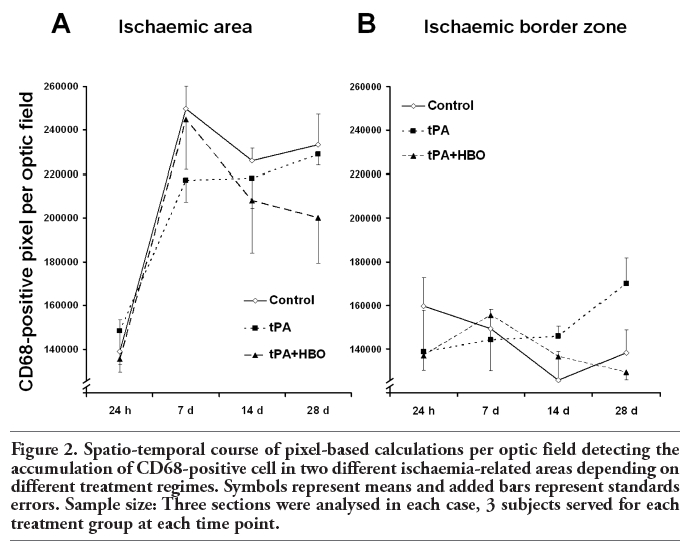 |
Figure 2. Spatio-temporal course of pixel-based calculations per optic field detecting the accumulation of CD68-positive cell in two different ischaemia-related areas depending on different treatment regimes. Symbols represent means and added bars represent standards errors. Sample size: Three sections were analysed in each case, 3 subjects served for each treatment group at each time point. |
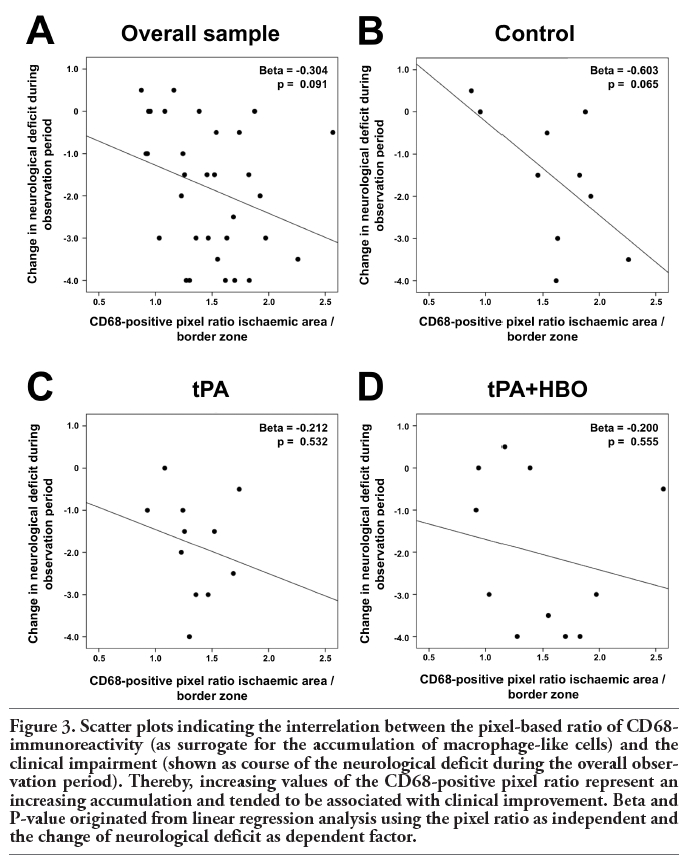 |
Figure 3. Scatter plots indicating the interrelation between the pixel-based ratio of CD68-immunoreactivity (as surrogate for the accumulation of macrophage-like cells) and the clinical impairment (shown as course of the neurological deficit during the overall observation period). Thereby, increasing values of the CD68-positive pixel ratio represent an increasing accumulation and tended to be associated with clinical improvement. Beta and P-value originated from linear regression analysis using the pixel ratio as independent and the change of neurological deficit as dependent factor. |
To obtain further insights into long-term reactions following acute focal cerebral ischaemia, triple immunofluorescence labelling was performed to address the co-occurrence of different inflammatory cell types. This approach was focussed on control animals at day 28, the group with most CD68-immunopositive cells in the present study (Figure 2A).
Figure 4 shows an ischaemic area with accumulation of macrophage-like cells (expressing CD68 and CD11b) and microglia (Iba). This region was clearly dominated by Iba- and CD11b-immunopositive cells appearing activated in the merged Figure 4D. Moreover, a few cells were stained by all three markers. Focussing on the border zone of focal cerebral ischaemia and taking astroglial reactions into account, Figure 5 demonstrates the concomitant staining of macrophage-like CD68-immunoreactive and Iba-positive microglial cells combined with the detection of GFAP-containing astroglia. In general, microglia and astroglia appeared activated. The boundary of accumulated macrophage-like cells was clearly pronounced, stretched from the left lower to the right upper corner in Figure 5A. Beyond this line, a strong astrogliosis was found in the ischaemic core-averted area, whereas a loss of astroglia occurred predominantly in the area of predominant macrophage-like accumulation. The regions of strongest Iba-labelling and highest accumulation of macrophage-like cells were largely identical, but activated microglia were also found in tissues devoid of CD68-staining. Additionally, considering the stroke-induced neuronal loss, Figure 6 represents the border zone from a case with an ischaemic lesion also affecting the neocortical tissue. Here, microgliosis was revealed by Iba staining (Figure 6A), whereas GFAP-immunoreactivity visualised astroglia (Figure 6B) and the neuronal loss was concomitantly detected by NeuN-immunolabelling (Figure 6C). The merged Figure 6D impressively displays the borderline of glial activation on the left side with simultaneous neuronal impairment. Finally, triple staining of Iba, GFAP and NeuN (Figure 7) exemplified the involvement of the hippocampal formation as region especially susceptible to hypoxia. Thereby, a strong microglial activation and astrocytic reaction occurred concurrently with diminished neurons in the CA1 region when focal ischaemia slightly, but not completely, affected the hippocampus.
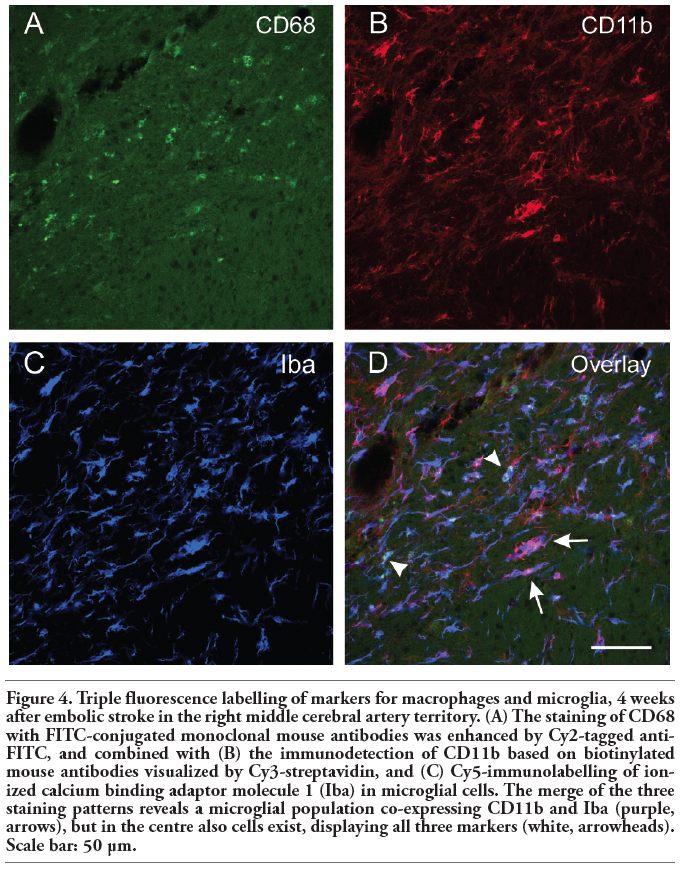 |
Figure 4. Triple fluorescence labelling of markers for macrophages and microglia, 4 weeks after embolic stroke in the right middle cerebral artery territory. (A) The staining of CD68 with FITC-conjugated monoclonal mouse antibodies was enhanced by Cy2-tagged anti-FITC, and combined with (B) the immunodetection of CD11b based on biotinylated mouse antibodies visualized by Cy3-streptavidin, and (C) Cy5-immunolabelling of ionized calcium binding adaptor molecule 1 (Iba) in microglial cells. The merge of the three staining patterns reveals a microglial population co-expressing CD11b and Iba (purple, arrows), but in the centre also cells exist, displaying all three markers (white, arrowheads). Scale bar: 50 µm. |
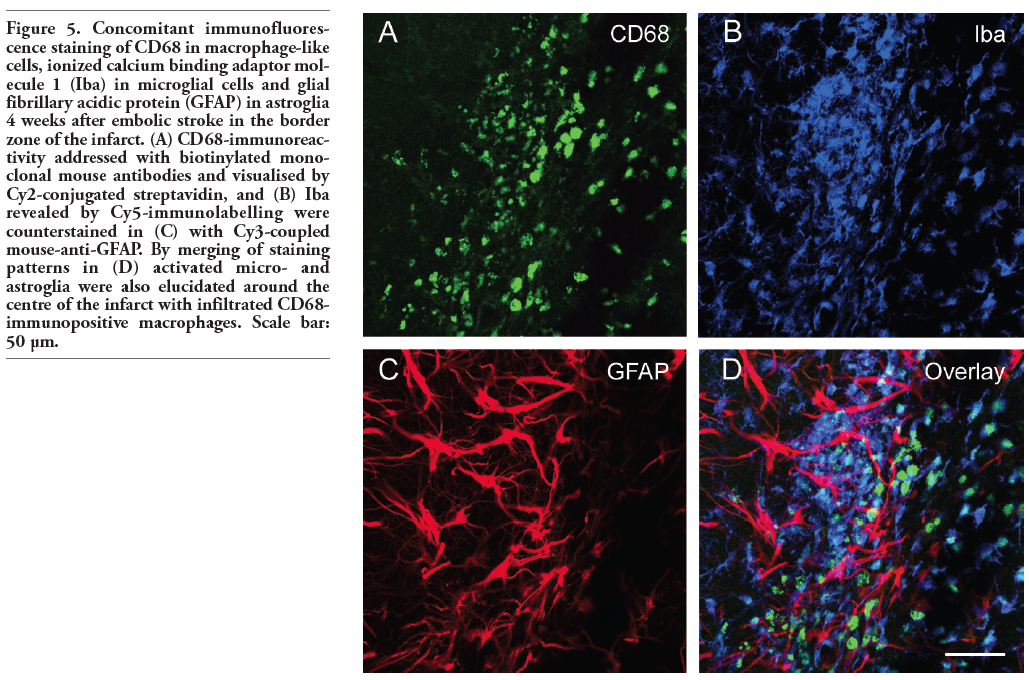 |
Figure 5. Concomitant immunofluorescence staining of CD68 in macrophage-like cells, ionized calcium binding adaptor molecule 1 (Iba) in microglial cells and glial fibrillary acidic protein (GFAP) in astroglia 4 weeks after embolic stroke in the border zone of the infarct. (A) CD68-immunoreactivity addressed with biotinylated monoclonal mouse antibodies and visualised by Cy2-conjugated streptavidin, and (B) Iba revealed by Cy5-immunolabelling were counterstained in (C) with Cy3-coupled mouse-anti-GFAP. By merging of staining patterns in (D) activated micro- and astroglia were also elucidated around the centre of the infarct with infiltrated CD68-immunopositive macrophages. Scale bar: 50 µm. |
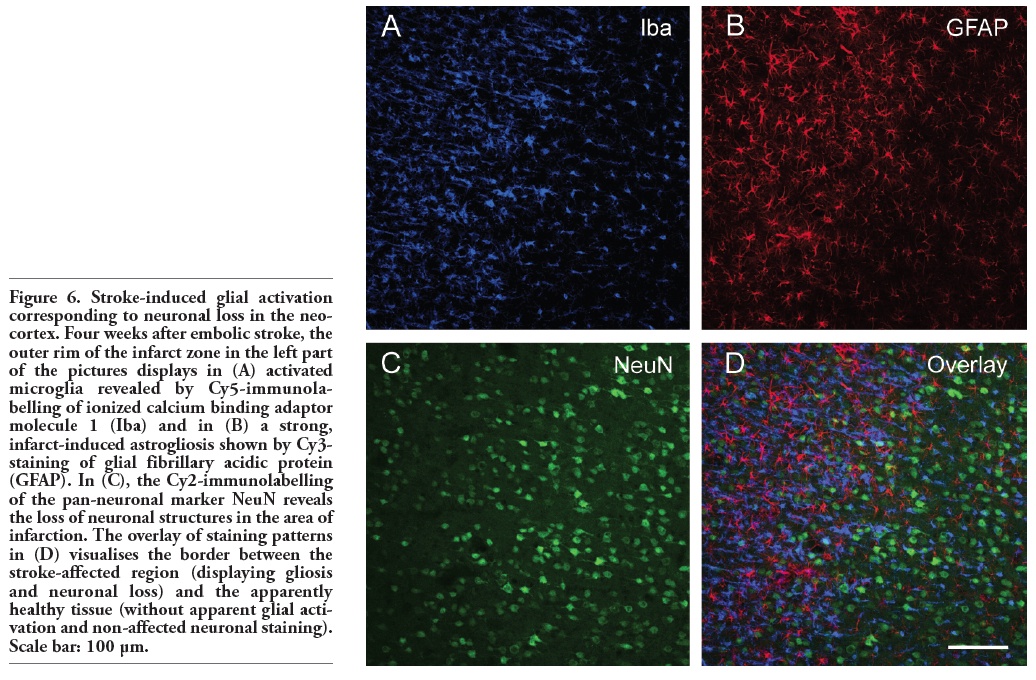 |
Figure 6. Stroke-induced glial activation corresponding to neuronal loss in the neocortex. Four weeks after embolic stroke, the outer rim of the infarct zone in the left part of the pictures displays in (A) activated microglia revealed by Cy5-immunolabelling of ionized calcium binding adaptor molecule 1 (Iba) and in (B) a strong, infarct-induced astrogliosis shown by Cy3-staining of glial fibrillary acidic protein (GFAP). In (C), the Cy2-immunolabelling of the pan-neuronal marker NeuN reveals the loss of neuronal structures in the area of infarction. The overlay of staining patterns in (D) visualises the border between the stroke-affected region (displaying gliosis and neuronal loss) and the apparently healthy tissue (without apparent glial activation and non-affected neuronal staining). Scale bar: 100 µm. |
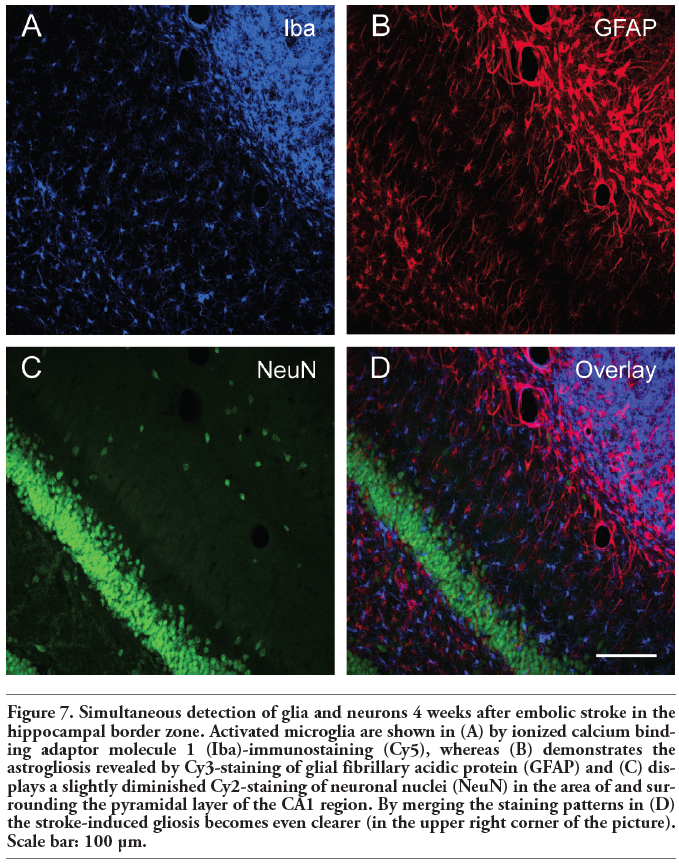 |
Figure 7. Simultaneous detection of glia and neurons 4 weeks after embolic stroke in the hippocampal border zone. Activated microglia are shown in (A) by ionized calcium binding adaptor molecule 1 (Iba)-immunostaining (Cy5), whereas (B) demonstrates the astrogliosis revealed by Cy3-staining of glial fibrillary acidic protein (GFAP) and (C) displays a slightly diminished Cy2-staining of neuronal nuclei (NeuN) in the area of and surrounding the pyramidal layer of the CA1 region. By merging the staining patterns in (D) the stroke-induced gliosis becomes even clearer (in the upper right corner of the picture). Scale bar: 100 µm. |
The present study addressed inflammatory reactions following focal cerebral ischaemia as a stroke research area with high priority,3 taking into account tPA as the only treatment currently promising in a clinical setting,23 and furthermore investigating HBO as potential neuroprotective co-medication to attenuate deleterious side effects of tPA.4,24,48,49 Special emphasis was given to a translational relevant experimental setting by precluding preconditioning attempts prior to stroke, using an embolic stroke model to mimic human conditions, and choosing a relatively long-term observation period of 4 weeks.1,4,62 Especially the latter feature of the present study achieves great relevance due to the known peak of inflammatory processes in later phases of stroke,7 which was neglected in experimental studies with shorter observation periods. Moreover, as conclusion from a study by Li et al. who demonstrated regionally different patterns of macrophages and neutrophils after MCAO,63 more than one ischaemia-related area was investigated.
In its first part, the present study reveals the spatio-temporal course of CD68-immunoreactivity as marker for the accumulation of macrophage-like cells, which has often been used in previous experimental stroke studies (e.g., Benjelloun et al.,58 and Qiao et al.64).
Focussing on the control group that represents the regular course of invading macrophage-like cells following focal cerebral ischaemia, a considerable increase was seen from 24 h to 7 days with long-lasting high levels up to day 28. In general, time-dependent changes have been summarised in several previous reports (e.g., Danton and Dietrich,9 Jin et al.,32 Stoll et al.13 and Tomita and Fukuuchi14), wherein, considering different influences of permanent vs transient ischaemia on post-stroke inflammation, an infiltration of leukocytes during the first hours and days has been described as ensured. Going into detail: in a notably comprehensive study, Clark et al. provided information about the development of infarct size and inflammatory mechanisms after permanent MCAO in rats and presented a model of 4 stages (neuronal death, inflammation, organization and resolution) within the first 30 days after onset of ischaemia.65 Additionally, stage II (inflammation) was subdivided into an initial/acute phase and a delayed/chronic phase, starting within the first 12 h, ending at day 5 and representing the infiltration of neutrophils; but abundant macrophages were also observed in stage III (organization), which ended at day 15.65 Comparable data of leukocyte invasion were provided, amongst others, by Kato et al. after transient MCAO in rats,66 Jander et al. and Kleinschnitz et al. after photochemically induced cortical ischaemia in rats,19,67 from Weston et al. after endothelin-caused MCAO in rats,68 and from Mena et al. who studied 137 post-mortem cases of human cerebral infarction.69 Typically, leukocyte invasion starts with a hypoxia-related production of inflammatory mediators by brain cells, which causes adhesion molecules being expressed on endothelial surfaces [e.g., the intercellular adhesion molecule 1 (ICAM-1)] thus enabling leukocyte infiltration.7,15,32,70,71 Several studies focussed on the roles of such invading cells: Gidday et al.,36 Justicia et al.,37 Romanic et al.,72 and Wang et al.38 demonstrated that leukocytes are a major source of MMP-9 following experimental stroke, and therefore contribute to the post-ischaemic blood-brain barrier disruption.26 These findings are in line with Rosell et al.,73 who confirmed the suggested association of neutrophil infiltration with local MMP-9 increase in patients after fatal cases of stroke. However, other reports indicate that MMPs might also be expressed by neurons.74 Predominantly deleterious effects were reported for MMPs in the acute phase following stroke, but recent studies also suggested regenerative properties of these enzymes in later phases.29,75 Concerning the maintenance of the inflammatory response after focal cerebral ischaemia, a recent study from Qiao et al. demonstrated that in contrast to mild ischaemia with only a transient inflammation, a long-lasting response up to weeks exists following a substantial infarction.64 This was supported by data from Karki et al.,76 who found a long-lasting presence of macrophages (up to 1 year) in the lesion border after transient MCAO in rats.
tPA was investigated in the present study due to its widespread use as a recanalisation drug with generally positive effects concerning the neurological outcome in acute stroke patients.23 Based on the knowledge that tPA activates also MMPs,25,27 which is even contributing to BBB impairment and increases the potential risk of haemorrhagic transformation,24,26 HBO was chosen to inhibit these side effects. This rationale was supported by reports from Liu et al. and Veltkamp et al.,77,78,79 who demonstrated reduced MMP-9 levels due to normobaric and hyperbaric hyperoxia after experimental stroke in rats. Furthermore, HBO was found to decrease the accumulation of neutrophils,45,46 possibly based on a HBO-related reduction of ICAM-1.80,81 In the present study, the accumulation of macrophage-like cells in the ischaemic area increases in a nearly identical way from 24 h to 7 days irrespective of treatment (see Figure 2A), which means that a combined application of HBO at 2.4 ata and tPA does not encompass a decreased inflammatory response during the first 7 days following experimental stroke. Similarly, Hjelde et al. reported neutrophil accumulation after permanent MCAO in rats following treatment with HBO at 2.0 ata or room air under ambient pressure.47 Focussing on the time course following day 7, a decreasing tendency of macrophage-like accumulation was observed in the tPA+HBO group.
Taking these results together with our previous work that showed a trend to worsened long-term clinical outcome (as defined by a combined endpoint of neurological deficit and premature death) in tPA+HBO-treated subjects vs the control and tPA group,50 the inhibition of macrophage-like accumulation in the ischaemic area has to be discussed in a critical manner. To further explore the general clinical impact of the obtained cellular changes, a functional perspective was added, addressing the relationship between the amount of macrophage-like cell accumulation and the reduction of neurological deficit during the overall observation period. Interestingly, these variables tended to be associated negatively, leading to the conclusion that an increased accumulation of macrophage-like cells might cause clinical improvement. Although it is important to note that the found association failed statistical significance, our data are in line with findings from a number of recent studies: Inácio et al. and Justicia et al. aimed to reduce the local inflammatory response after cerebral ischaemia via antibodies recognising the vascular cell adhesion molecule-1 or via an inhibition of macrophage migration while using factor-depleted mice.82,83 Furthermore, Shechter et al. controlled macrophage invasion after spinal cord injury.84 Taken together, these attempts did not lead to beneficial effects. In parallel, Madinier et al. reported that the drug-induced reduction of the post-ischaemic inflammatory response, especially the microglia activation, causes a long-term impairment of neuronal plasticity.85 Moreover, Smirkin et al. and Matsumoto et al. identified populations of invading cells in the ischaemic area after transient MCAO in rats playing regenerative roles, e.g., by producing neuroprotective factors.86,87 Finally, Manoonkitiwongsa et al. found a strong association between the microvessel density and the macrophage count in relation to impaired brain tissue after transient MCAO in rats and concluded that these microvessels enable the infiltration of macrophages, compatible to the clean-up hypothesis.88
The second part of the present study was focussed on the attempt to differentiate delayed components of the inflammatory response, known to be highly complex.8,9,13,14,89,90 Additionally, neurons were addressed due to their embedding in the neurovascular unit concept.5,6,10 Microglial and astroglial activation was described by numerous reports following cerebral ischaemia indicating their role as key elements.13,28,32,55,65,66,91,92,93 In this regard, recent studies elucidated microglia as a source of pro-inflammatory mediators such as tumor necrosis factor in early stages of ischaemia.32,33,94,95,96 However, neuroprotective properties of microglia have also been discussed.12,28,32,97,98,99,100
Summarising data from the present study, a strong microglial activation was still found at 4 weeks following focal cerebral ischaemia, indicating that microglia play not only a role in early phases following stroke. This activation was mainly co-localised with macrophage-like cells as indicated by CD68. Interestingly, at this time point the general microglia activation resulting in altered cell morphology appears as dominant effect when compared with the occurrence of CD68-positive cells, even if some Iba/CD68-co-expressing cells exist. Our findings are in line with a previous report by Gelderblom et al. who investigated the time course of post-stroke inflammation up to 7 days after MCAO in mice and found a larger proportion of microglia vs macrophages in this interval.101 Furthermore, the present data are in accordance with the reported microglial activation observed even 4 weeks after MCAO.102 The activated microglia were thereby primary associated to the area of ischaemia-related impairment as shown by neuronal loss, which was described previously in a comparable manner but with markedly shorter observation periods.71,93,103 Focussing on astrocytes, at 4 weeks a strong circular astrogliosis was found in the rim around CD68-positive cells (see Figures 5 and 6) representing the border zone of ischaemia enwrapping the infarct core. Such an organization has been described previously (e.g., by Verkhratsky and Butte104), whereas Clark et al. investigated this issue in a long-term course and found that this process starts to form up at 24 h after MCAO simultaneously to neutrophil infiltration and lasts up to day 30, enabling the demarcation of the necrotic core.65
The relatively small number of subjects limits the validity of the study. However, the per-formed semiquantitative evaluation allows a tendency of spatio-temporal alterations for macrophage-like cells after experimental embolic stroke, which is of high relevance regarding translational issues. The found long-term course towards a decreased macrophage-like cell accumulation in the combined treatment group (tPA+HBO) is presumably related to the co-application of HBO, but due to the lack of a separate HBO group the definitive proof remains open. Caused by the overlapping target structures of available antibodies in this field, it represents an ongoing challenge to address neural subpopulations specifically, e.g., for the clarification on proportional invading macrophages, differentiated from activated resident microglia, as addressed by previous reviews (e.g., Dijkstra et al.,105 Matsumoto et al.,18 and Stoll et al.13). Especially for microglia, it remains uncertain if the obtained circular organization originates from proliferation of resident microglia or invading processes. In the past, several lectins (e.g., from Griffonia simplicifolia106) were useful markers for microglia but co-stained vessels,57,107 which was not desired in the present study.
This study reveals a spatio-temporal pattern of accumulating macrophage-like cells after experimental ischaemic stroke with a significant increase from 24 h to 7 days post ischaemia, which remained up to 4 weeks, indicating a long-term inflammatory response. Taking into account tPA as a widely used recanalisation drug and HBO a as potential neuroprotective approach, a tendency in the combined treatment group to a decreased accumulation of macrophage-like cells starting at day 14 needs to be discussed in a critical manner, since recent research has ascribed long-term inflammatory responses as important for repair and neuronal plasticity. Following experimental stroke, a long-lasting glial activation was found simultaneously with neuronal loss. Thereby, microglia were mainly observed within the ischaemic area whereas astrogliosis occurred primarily in the ischaemic border zone.
1. Endres M, Engelhardt B, Koistinaho J, Lindvall O, Meairs S, Mohr JP, et al. Improving outcome after stroke: overcoming the translational roadblock. Cerebrovasc Dis 2008;25:268-78. [CrossRef][PubMed]
2. Diener HC, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, et al. NXY-059 for the treatment of acute stroke: pooled analysis of the SAINT I and II Trials. Stroke 2008;39:1751-8.[CrossRef][PubMed]
3. Meairs S, Wahlgren N, Dirnagl U, Lindvall O, Rothwell P, Baron JC, et al. Stroke research priorities for the next decade-A representative view of the European scientific community. Cerebrovasc Dis 2006;22: 75-82.[CrossRef][PubMed]
4. Young AR, Ali C, Duretête A, Vivien D. Neuroprotection and stroke: time for a compromise. J Neurochem 2007;103:1302-9.[CrossRef][PubMed]
5. Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol 2007;28:5-11.[CrossRef][PubMed]
6. del Zoppo GJ. Relationship of neurovascular elements to neuron injury during ischemia. Cerebrovasc Dis 2009;27 (Suppl.1):65-76.[CrossRef][PubMed]
7. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999;22:391-7.[CrossRef][PubMed]
8. Mergenthaler P, Dirnagl U, Meisel A. Pathophysiology of stroke: lessons from animal models. Metab Brain Dis 2004;19: 151-67.[CrossRef][PubMed]
9. Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 2003;62:127-36. [PubMed]
10. del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009;158: 972-82.[CrossRef][PubMed]
11. Gehrmann J, Bonnekoh P, Miyazawa T, Hossmann KA, Kreutzberg GW. Immunocytochemical study of an early microglial activation in ischemia. J Cereb Blood Flow Metab 1992;12:257-69.[CrossRef]
12. Lees GJ. The possible contribution of microglia and macrophages to delayed neuronal death after ischemia. J Neurol Sci 1993;114:119-22.[CrossRef][PubMed]
13. Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 1998;56:149-71.[CrossRef][PubMed]
14. Tomita M, Fukuuchi Y. Leukocytes, macrophages and secondary brain damage following cerebral ischemia. Acta Neurochir Suppl 1996;66:32-9.[PubMed]
15. Tuttolomondo A, Di Sciacca R, Di Raimondo D, Renda C, Pinto A, Licata G. Inflammation as a therapeutic target in acute ischemic stroke treatment. Curr Top Med Chem 2009;9:1240-60.[CrossRef][PubMed]
16. Hulette CM, Downey BT, Burger PC. Macrophage markers in diagnostic neuropathology. Am J Surg Pathol 1992;16: 493-9.[CrossRef][PubMed]
17. Krupinski J, Kaluza J, Kumar P, Kumar S. Immunocytochemical studies of cellular reaction in human ischemic brain stroke. MAB anti-CD68 stains macrophages, astrocytes and microglial cells in infarcted area. Folia Neuropathol 1996;34:17-24.[PubMed]
18. Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Li C, et al. Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions. J Neurosci Res 2007;85:994-1009.[CrossRef][PubMed]
19. Kleinschnitz C, Bendszus M, Frank M, Solymosi L, Toyka KV, Stoll G. In vivo monitoring of macrophage infiltration in experimental ischemic brain lesions by magnetic resonance imaging. J Cereb Blood Flow Metab 2003;23:1356-61.[CrossRef][PubMed]
20. Wiart M, Davoust N, Pialat JB, Desestret V, Moucharrafie S, Cho TH, et al. MRI monitoring of neuroinflammation in mouse focal ischemia. Stroke 2007;38:131-7.[CrossRef][PubMed]
21. Saleh A, Schroeter M, Jonkmanns C, Hartung HP, Mödder U, Jander S. In vivo MRI of brain inflammation in human ischaemic stroke. Brain 2004;127:1670-7.[CrossRef][PubMed]
22. Orset C, Haelewyn B, Vivien K, Vivien D, Young AR. Rodent models of thromboembolic stroke. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press c/o Springer Science + Business Media; 2010. p. 55-70.
23. Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008;359: 1317-29.[CrossRef][PubMed]
24. Adibhatla RM, Hatcher JF. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: therapeutic strategies. CNS Neurol Disord Drug Targets 2008;7:243-53.[CrossRef][PubMed]
25. Burggraf D, Martens HK, Dichgans M, Hamann GF. rt-PA causes a dose-dependent increase in the extravasation of cellular and non-cellular blood elements after focal cerebral ischemia. Brain Res 2007;1164:55-62.[CrossRef][PubMed]
26. Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis 2010;38:376-85.[CrossRef][PubMed]
27. Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol 2006;200:38-49.[CrossRef][PubMed]
28. Jordán J, Segura T, Brea D, Galindo MF, Castillo J. Inflammation as therapeutic objective in stroke. Curr Pharm Des 2008; 14:3549-64.[CrossRef][PubMed]
29. Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol 2008;8:82-9.[CrossRef][PubMed]
30. Giulian D, Corpuz M, Chapman S, Mansouri M, Robertson C. Reactive mononuclear phagocytes release neurotoxins after ischemic and traumatic injury to the central nervous system. J Neurosci Res 1993;36:681-93.[CrossRef][PubMed]
31. Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 2000;20: 53-65.[CrossRef][PubMed]
32. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010;87:779-89.[CrossRef][PubMed]
33. Lambertsen KL, Clausen BH, Fenger C, Wulf H, Owens T, Dagnaes-Hansen F, et al. Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neuroscience 2007;144:934-49. [CrossRef][PubMed]
34. Lindsberg PJ, Strbian D, Karjalainen-Lindsberg ML. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J Cereb Blood Flow Metab 2010;30:689-702. [CrossRef][PubMed]
35. Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005;50:329-39.[CrossRef][PubMed]
36. Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol 2005;289:H558-68.[CrossRef][PubMed]
37. Justicia C, Panés J, Solé S, Cervera A, Deulofeu R, Chamorro A, et al. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab 2003;23:1430-40.[CrossRef][PubMed]
38. Wang G, Guo Q, Hossain M, Fazio V, Zeynalov E, Janigro D, et al. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Wang G, Guo Q, Hossain M, Fazio V, Zeynalov E, Janigro D, et al. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res 2009;1294:183-92.[CrossRef][PubMed]
39. Calvert JW, Cahill J, Zhang JH. Hyperbaric oxygen and cerebral physiology. Neurol Res 2007;29:132-41.[CrossRef][PubMed]-
40. Jain KK. Textbook of Hyperbaric Medicine. 5th revised and updated edition. Göttingen: Hogrefe & Huber; 2009. [PubMed]
41. Henninger N, Küppers-Tiedt L, Sicard KM, Günther A, Schneider D, Schwab S. Neuroprotective effect of hyperbaric oxygen therapy monitored by MR-imaging after embolic stroke in rats. Exp Neurol 2006;201:316-23.[CrossRef][PubMed]
42. Schäbitz WR, Schade H, Heiland S, Kollmar R, Bardutzky J, Henninger N, et al. Neuroprotection by hyperbaric oxygenation after experimental focal cerebral ischemia monitored by MRI. Stroke 2004;35:1175-9.[CrossRef][PubMed]
43. Veltkamp R, Warner DS, Domoki F, Brinkhous AD, Toole JF, Busija DW. Hyperbaric oxygen decreases infarct size and behavioral deficit after transient focal cerebral ischemia in rats. Brain Res 2000; 853:68-73.[CrossRef][PubMed]
44. Sun L, Zhou W, Mueller C, Sommer C, Heiland S, Bauer AT, et al. Oxygen therapy reduces secondary hemorrhage after thrombolysis in thromboembolic cerebral ischemia. J Cereb Blood Flow Metab 2010; 30:1651-60.[CrossRef][PubMed]
45. Miljkovic-Lolic M, Silbergleit R, Fiskum G, Rosenthal RE. Neuroprotective effects of hyperbaric oxygen treatment in experimental focal cerebral ischemia are associated with reduced brain leukocyte myeloperoxidase activity. Brain Res 2003; 971:90-4.[CrossRef][PubMed]
46. Atochin DN, Fisher D, Demchenko IT, Thom SR. Neutrophil sequestration and the effect of hyperbaric oxygen in a rat model of temporary middle cerebral artery occlusion. Undersea Hyperb Med 2000;27: 185-90.[PubMed]
47. Hjelde A, Hjelstuen M, Haraldseth O, Martin D, Thom R, Brubakk O. Hyperbaric oxygen and neutrophil accumulation/tissue damage during permanent focal cerebral ischaemia in rats. Eur J Appl Physiol 2002;86:401-5.[CrossRef][PubMed]
48. Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res 2007;29:173-83.[CrossRef][PubMed]
49. Michalski D, Härtig W, Schneider D, Hobohm C. Use of normobaric and hyperbaric oxygen in acute focal cerebral ischemia - a preclinical and clinical review. Acta Neurol Scand 2011;123:85-97.[CrossRef][PubMed]
50. Michalski D, Küppers-Tiedt L, Weise C, Laignel F, Härtig W, Raviolo M, et al. Long-term functional and neurological outcome after simultaneous treatment with tissue-plasminogen activator and hyperbaric oxygen in early phase of embolic stroke in rats. Brain Res 2009;1303:161-8.[CrossRef][PubMed]
51. Zhang RL, Chopp M, Zhang ZG, Jiang Q, Ewing JR. A rat model of focal embolic cerebral ischemia. Brain Res 1997;766:83-92.[CrossRef][PubMed]
52. Härtig W, Brückner G, Holzer M, Brauer K, Bigl V. Digoxigenylated primary antibodies for sensitive dual-peroxidase labelling of neuronal markers. Histochem. Cell Biol 1995;104:467-72.[CrossRef][PubMed]
53. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387-94. [CrossRef][PubMed]
54. Hanisch UK, van Rossum D. Microglia properties. Encyclopedia of Neuroscience 2009;5:853-9.
55. Lehrmann E, Christensen T, Zimmer J, Diemer NH, Finsen B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. J Comp Neurol 1997;386:461-76.[CrossRef][PubMed]
56. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003;183:25-33.[CrossRef][PubMed]
57. Härtig W, Reichenbach A, Voigt C, Boltze J, Bulavina L, Schuhmann MU, et al. Triple fluorescence labelling of neuronal, glial and vascular markers revealing pathological alterations in various animal models. J Chem Neuroanat 2009;37:128-38.[CrossRef][PubMed]
58. Benjelloun N, Renolleau S, Represa A, Ben-Ari Y, Charriaut-Marlangue C. Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal rat. Stroke 1999;30:1916-23.[CrossRef][PubMed]
59. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 1998;57:1-9. [CrossRef]
60. Weiss LM, Arber DA, Chang KL. CD68. A Review. Appl Immunohistochem 1994;2:2-8.
61. Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992;116:201-11.[PubMed]
62. Li F, Tatlisumak T. Focal brain ischemia models in rodents. In: Tatlisumak T, Fisher M, editors. Handbook of Experimental Neurology. Cambridge: Cambridge University Press; 2006. p. 212-28.[CrossRef]
63. Li Y, Powers C, Jiang N, Chopp M. Intact, injured, necrotic and apoptotic cells after focal cerebral ischemia in the rat. J Neurol Sci 1998;156:119-32.[CrossRef][PubMed]
64. Qiao M, Meng S, Foniok T, Tuor UI. Mild cerebral hypoxia-ischemia produces a sub-acute transient inflammatory response that is less selective and prolonged after a substantial insult. Int J Dev Neurosci 2009;27:691-700.[CrossRef][PubMed]
65. Clark RK, Lee EV, Fish CJ, White RF, Price WJ, Jonak ZL, et al. Development of tissue damage, inflammation and resolution following stroke: an immunohistochemical and quantitative planimetric study. Brain Res Bull 1993;31:565-72.[CrossRef][PubMed]
66. Kato H, Kogure K, Liu XH, Araki T, Itoyama Y. Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal cerebral ischemia in the rat. Brain Res 1996;734: 203-12.[CrossRef][PubMed]
67. Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab 1995;15:42-51.[CrossRef][PubMed]
68. Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab 2007;27: 100-14.[CrossRef][PubMed]
69. Mena H, Cadavid D, Rushing EJ. Human cerebral infarct: a proposed histopathologic classification based on 137 cases. Acta Neuropathol 2004;108:524-30.[CrossRef][PubMed]
70. Lindsberg PJ, Carpén O, Paetau A, Karjalainen-Lindsberg ML, Kaste M. Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation 1996;94:939-45.[CrossRef][PubMed]
71. Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol 1994;55:195-203.[CrossRef][PubMed]
72. Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998;29:1020-30.[CrossRef][PubMed]
73. Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008;39:1121-6.[CrossRef][PubMed]
74. Planas AM, Solé S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischaemia. Neurobiol Dis 2001;8:834-46.[CrossRef][PubMed]
75. Morancho A, Rosell A, García-Bonilla L, Montaner J. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci 2010;1207: 123-33.[CrossRef][PubMed]
76. Karki K, Knight RA, Shen LH, Kapke A, Lu M, Li Y, et al. Chronic brain tissue remodeling after stroke in rat: a 1-year multiparametric magnetic resonance imaging study. Brain Res 2010;1360:168-76.[CrossRef][PubMed]
77. Liu W, Hendren J, Qin XJ, Liu KJ. Normobaric hyperoxia reduces the neurovascular complications associated with delayed tissue plasminogen activator treatment in a rat model of focal cerebral ischemia. Stroke 2009;40:2526-31.[CrossRef][PubMed]
78. Liu W, Hendren J, Qin XJ, Shen J, Liu KJ. Normobaric hyperoxia attenuates early blood-brain barrier disruption by inhibiting MMP-9-mediated occludin degradation in focal cerebral ischemia. J Neurochem 2009;108:811-20.[CrossRef][PubMed]
79. Veltkamp R, Bieber K, Wagner S, Beynon C, Siebing DA, Veltkamp C, et al. Hyperbaric oxygen reduces basal lamina degradation after transient focal cerebral ischemia in rats. Brain Res 2006;1076:231-7.[CrossRef][PubMed]
80. Buras JA, Reenstra WR. Endothelial-neutrophil interactions during ischemia and reperfusion injury: basic mechanisms of hyperbaric oxygen. Neurol Res 2007;29: 127-31.[CrossRef][PubMed]
81. Hong JP, Kwon H, Chung YK, Jung SH. The effect of hyperbaric oxygen on ischemia-reperfusion injury: an experimental study in a rat musculocutaneous flap. Ann Plast Surg 2003;51:478-87.[CrossRef][PubMed]
82. Inácio AR, Ruscher K, Leng L, Bucala R, Deierborg T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J Cereb Blood Flow Metab 2011;31:1093-106.[CrossRef][PubMed]
83. Justicia C, Martín A, Rojas S, Gironella M, Cervera A, Panés J, et al. Anti-VCAM-1 antibodies did not protect against ischemic damage either in rats or in mice. J Cereb Blood Flow Metab 2006;26:421-32.[CrossRef][PubMed]
84. Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 2009;6:e1000113.[CrossRef][PubMed]
85. Madinier A, Bertrand N, Mossiat C, Prigent-Tessier A, Beley A, Marie C, et al. Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS One 2009;4:e8101.[CrossRef][PubMed]
86. Smirkin A, Matsumoto H, Takahashi H, Inoue A, Tagawa M, Ohue S, et al. Iba1+/NG2+ macrophage-like cells expressing a variety of neuroprotective factors ameliorate ischemic damage of the brain. J Cereb Blood Flow Metab 2010;30:603-15.[CrossRef][PubMed]
87. Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Chuai M, et al. Accumulation of macrophage-like cells expressing NG2 proteoglycan and Iba1 in ischemic core of rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 2008;28:149-63.[CrossRef][PubMed]
88. Manoonkitiwongsa PS, Jackson-Friedman C, McMillan PJ, Schultz RL, Lyden PD. Angiogenesis after stroke is correlated with increased numbers of macrophages: the clean-up hypothesis. J Cereb Blood Flow Metab 2001;21:1223-31.[CrossRef][PubMed]
89. Brouns R, DeDeyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg 2009;111:483-95.[CrossRef][PubMed]
90. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009;15:192-9.[CrossRef][PubMed]
91. Hobohm C, Günther A, Grosche J, Rossner S, Schneider D, Brückner G. Decomposition and long-lasting downregulation of extracellular matrix in perineuronal nets induced by focal cerebral ischemia in rats. J Neurosci Res 2005;80:539-48.[CrossRef][PubMed]
92. Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 1997;20:570-7. [CrossRef][PubMed]
93. Weinstein JR, Koerner IP, Möller T. Microglia in ischemic brain injury. Future Neurol 2010;5:227-46.[CrossRef][PubMed]
94. Halleskog C, Mulder J, Dahlström J, Mackie K, Hortobágyi T, Tanila H, et al. WNT signaling in activated microglia is proinflammatory. Glia 2011;59:119-31.[CrossRef][PubMed]
95. Lambertsen KL, Meldgaard M, Ladeby R, Finsen B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J Cereb Blood Flow Metab 2005;25:119-35.[CrossRef][PubMed]
96. Nakamura Y. Regulating factors for microglial activation. Biol Pharm Bull 2002;25:945-53.[CrossRef][PubMed]
97. Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 2007;27:1941-53.[CrossRef][PubMed]
98. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996;19:312-8.[CrossRef][PubMed]
99. Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematol Disord 2004;4:65-84.[CrossRef][PubMed]
100. Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002;40:133-9.[CrossRef][PubMed]
101. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009;40:1849-57.[CrossRef][PubMed]
102. Morioka T, Kalehua AN, Streit WJ. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J Comp Neurol 1993;327:123-32.[CrossRef][PubMed]
103. Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y, Yanagihara T, et al. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 2000;31:1735-43.[CrossRef][PubMed]
104. Verkhratsky A, Butte A. Glia and Diseases of the nervous system. In: Verkhratsky A, Butte A, editors. Glial Neurobiology. A Textbook. Chichester: John Wiley & Sons Ltd; 2007. p. 167-96.[CrossRef]
105. Dijkstra CD, Döpp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology 1985;54:589-99.[PubMed]
106. Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4). J Histochem Cytochem 1990;38:1683-6.[CrossRef][PubMed]
107. Acarin L, Vela JM, González B, Castellano B. Demonstration of poly-N-acetyl lactosamine residues in ameboid and ramified microglial cells in rat brain by tomato lectin binding. J Histochem Cytochem 1994;42:1033-41. [CrossRef][PubMed]
[TOP]